NON-CONFIDENTIAL PATENT DESCRIPTION
"Computational Analysis of Information in Genetic Diagnosis and Design of Genetic Control Elements"
By P.K. Rogan and T.D. Schneider
PSU Invention Disclosure No. 94-1440
(US Ser. No. PCT/US96/11088)
This patent was allowed in July 1997
This invention relates to a method for identifying and manipulating the binding affinity of a particular position within and surrounding a binding site. The instant method allows comparison of the information on particular binding sites to the individual information content of other binding sites, to distances between features of the sequence, and to their measured binding energies. Software allows the user to investigate the effects of sequence changes in the regions around the binding site to detect the creation or destruction of nearby binding sites.
One application of the present invention involves adjustment of the affinity of a binding site by manipulating positions within the site to alter its individual information content. Specific applications include the design of genetic control elements of the strength required for a particular application or the active site in an enzyme or other motifs in proteins and drug binding sites.
The invention also permits the prediction of whether the nucleotide substitutions in a DNA sequence will be a deleterious mutation or will be benign. Information analysis produces a quantitative measure of sequence conservation for a set of sequences with similar function and the test sequence carrying the nucleotide substitution. The information in the test sequence is compared to the distribution of information contents derived from a conserved set of functional DNA sequences. This computational method requires no experimental analysis aside from determining the sequence bearing the nucleotide substitution.
Of the 105 nucleotide substitutions examined initially, polymorphic changes in splice sites were identified that were presumed in the original reports to be mutations that alter splice efficiency or the sequence of the mature mRNA. These included nucleotide changes in the familial non-polyposis colon cancer gene MSH2 (Rogan and Schneider, Human Mutation, 6: 74-76, 1995), the p53 gene which has been associated with some instances of bladder carcinoma, the gene encoding ornithine-transcarbamylase, and the gene encoding steroid 21-hydroxylase causing adrenal hyperplasia. This technique also detected cryptic splice sites that were not apparent in the original reports. The misidentification of such mutations may have important clinical and legal implications. This method can be applied to the analysis of any amino acid or nucleotide substitution in a conserved sequence element of any type.
Both U.S. and foreign patent applications are currently pending for this invention disclosure. The software and the methodology for design of genetic control elements and genetic diagnostics are currently available for licensing. Exclusive, nonexclusive, internal commercial use, and commercial evaluation licensing opportunities are available for this invention.
FOR MORE
INFORMATION, PLEASE CONTACT US
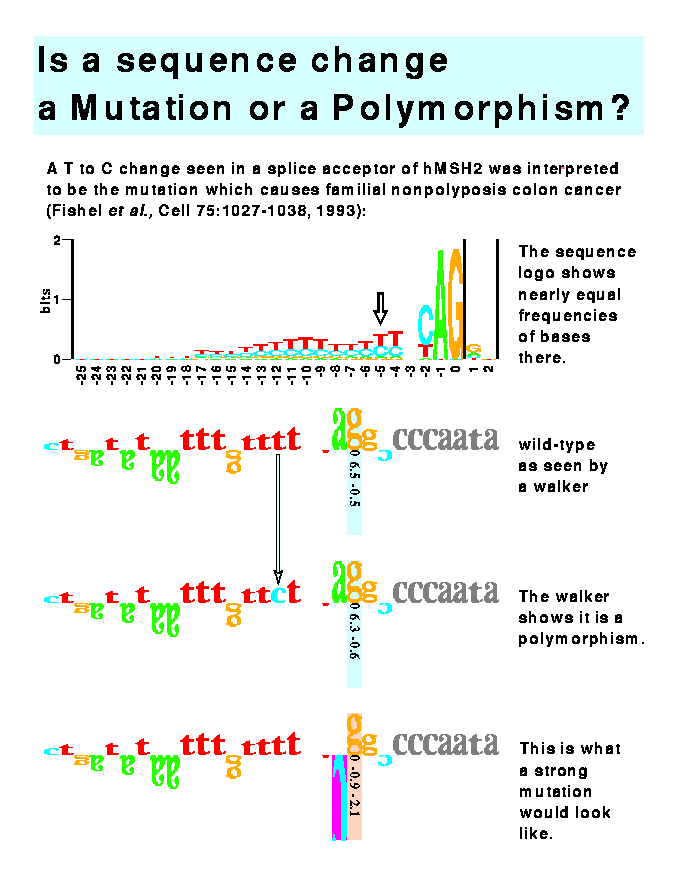
LEGEND TO THE FIGURE
This figure demonstrates the use of the mutation feature of the software program to distinguish mutations from polymorphic changes. Three rows of sequence are given. These represent modifications of one sequence. The top sequence in Figure 10 is the human splice acceptor site given in Fishel et al. (1993. Cell, 75:1027-1038). This is the DNA sequence found in normal colon tissue. The middle sequence is that found in a sporadic colorectal tumor. Fishel et al. proposed that this T-C change at position -5 was the cause of the cancer, but the software immediately shows that this change is not significant since the individual information content (Ri) only changes from 6.5 to 6.3 bits and the absolute value of the Z score is still below 1. Thus, this change represents a polymorphism and not a mutation. The true mutation lies elsewhere or this mutation represents a change in the binding site for some molecule other than the spliceosome. The bottom row shows the effect of altering the sequence in the top row: when position -1 is changed to a cytosine ("C"), the Ri becomes negative and the Z score approaches significance (p<0.02). Such an alteration would probably lead to colon cancer.

Poster
on Individual Information.
origin: 1996 Jul 25
updated: 2012 Apr 11