How Can I Make
Sequence Logos
on My Own Computer?
There are two ways to make sequence logos,
the Alpro Route
and
the Delila Route.
If you are interested in proteins
or your sequence has gaps then the
you must use the Alpro Route.
If you are working
with binding sites, the Delila Route has great advantages.
FOR BOTH ROUTES
To use the programs, you must first set them up
on your computer.
The programs are written in Pascal and either you
can get a binary (if you work on a Sun machine) or
you need to compile them.
The currently available methods are
described on the
Delila Software page.
Documentation, source code and Sun binaries of
each program are given on the pages linked to below.
First try the
test.p
program. If you can get this program to run, many other programs will work.
Then get
delmod.p
and run it. This tests the date/time feature, which is system
dependent. If it works ok, all programs should be ok.
For more tips see
Setting Up and Using Delila Programs.
To learn how the Delila system is designed,
see
LIBDEF,
the definition of the Delila system.
ALPRO ROUTE: proteins
or nucleic acid binding sites
If you have alignments with gaps or protein sequences, then you only
need (and must use)
alpro
and
makelogo.
The steps are:
- Prepare the protseq file and then run
alpro.
-
Run
makelogo.
DELILA ROUTE: nucleic acid binding sites
If you are working with binding sites
in GenBank, you will need many programs. They are given below in the order
you use them, with a brief explanation of each.
Be sure to read the manual
page of each one.
In particular, note how the output of each program generally becomes the input
to the next.
Also, it helps to read various definitions in the
glossary.
- dbbk: Convert GenBank flat file format to Delila format,
which is called
a `book' (db = database, bk = book).
This produces an 'l1' file which contains the book.
(l1 is a lower case L [standing for 'Library'] followed by the symbol
'one').

- catal: Catalogue the contents of a book, usually l1.
If you are not using them (which is generally the case)
make empty l2, l3 and catalp files, then run catal.
This will produce six files:
lib1, lib2, lib3 and
cat1, cat2, cat3.
These make up the library used by Delila in the next step.
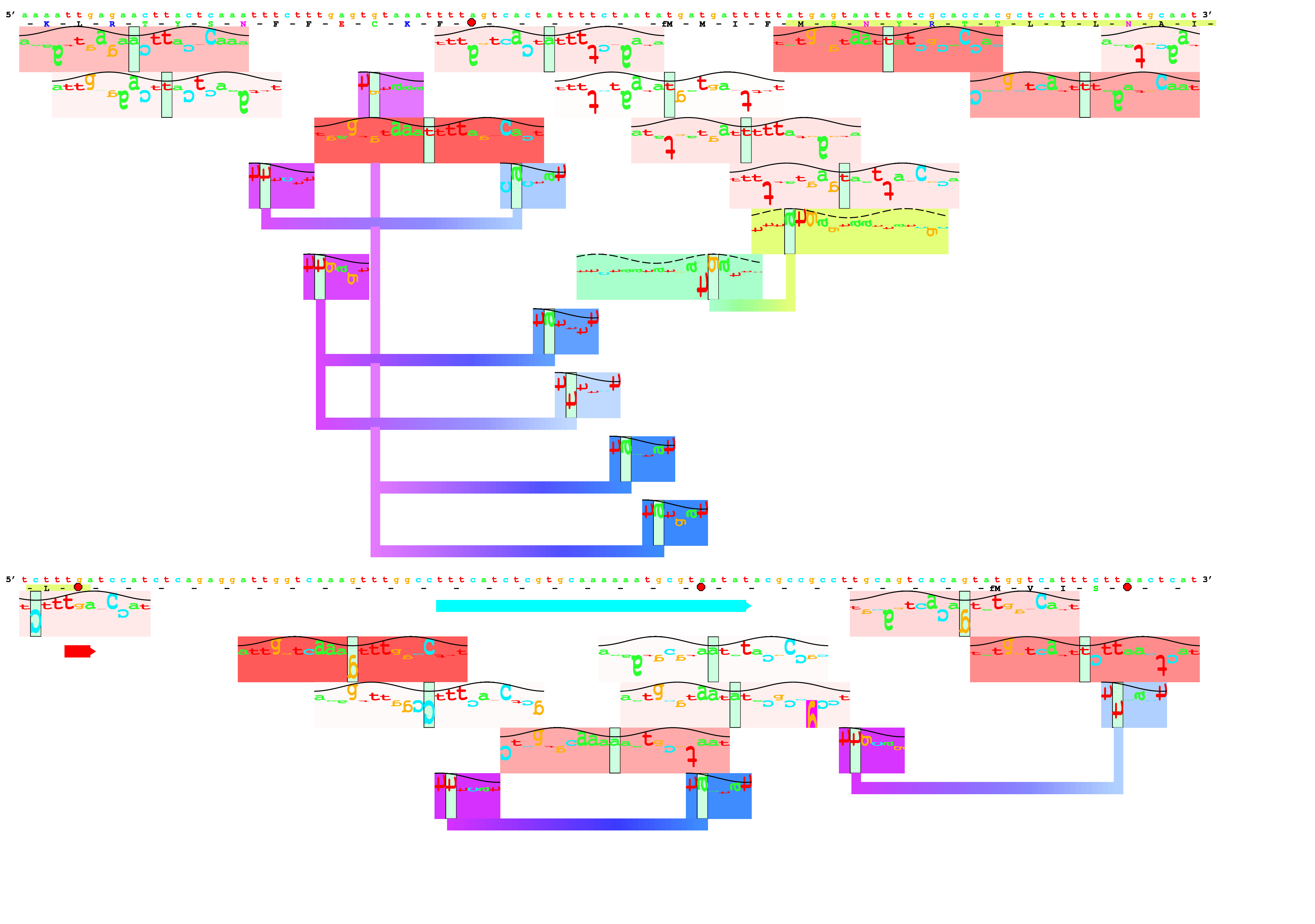
- delila: Extract fragments of sequences from a library of sequences
and create a subset, a book. This is the core of the Delila system.
You give Delila instructions (inst file) and those are used to create the subset
desired. For a logo, generally one makes instructions that look like this:
get from 5600 -200 to same +200 direction +;
This means to get 200 bases before position 5600 to 200 bases afterwards,
for a total of 401 bases.
I recommend that you use a wide
range like this to be able to see the background noise
around the binding site.
By the way, the number 5600 becomes the
zero coordinate of the
binding site.
I try to pick a position that is strongly conserved
(high information content).
See the
Introduction to Delila Instructions for more details.
- alist: Make an aligned listing of sequences using the Delila instructions
and the book created by Delila.
The pair of the book and inst file is a set called an 'aligned book'.
I create a listing of the
sequences to be sure that they are aligned correctly.
If this works, making the logo is fast and a piece of cake
(or slice pizza ;-)
Even if you have a large
range defined in the delila instructions, you can use
a smaller one for the aligned listing. Note that running this
program does not affect the inst or book files, so can't affect
later steps.
The program is controled by a file `alistp', which stands
for alist-parameters.
See the glossary definition of
parameter file.
- encode: Convert the book/inst into 0 and 1's.
This is historically the way we did it, but it is fast
so we still do it this way. The output is encseq, which is a bit large.
To save space, you can delete the encseq file after running the next program.
Note that the
range
can be reduced in the parameter file, so I normally
set this to -200 to 200.
- rseq: Compute
Rsequence
for each position in the aligned book,
to make an rsdata file.
- dalvec: Convert the rsdata file into a symvec file, which the next
program can use to make the logo.
- makelogo: Finally! Make the
sequence logo!
There are lots of parameters you can adjust.
- rf: Once you have computed the information content of your binding
site, you may want to compute
Rfrequency.
You will need to know how big the genome is (or the total number
of binding positions availble to the
recognizer)
and the number of
binding sites in that region.
Displaying the Logo
- PostScript: Give the logo file to a postscript printer or display
device and you will have the logo.
A good postscript display is ghostview.
- Convert to PDF. However, now-a-days I convert to PDF
using my
showpdf script
(which depends on
ps2pdf
available by
fink
and my
ish script).
I then look at the PDF with
Acrobat
or
Skim.
The advantage of Skim is that it can be set to watch the logo file
and update when that changes using
atchange.
This way one can edit the parameter file and see the logo
change without moving one's fingers from the keyboard.
After the Logo: Individual Information
Once you have a sequence logo, it is only one step to create
the
individual information
model and to create
sequence walkers.
All of these programs are part of the
Individual Information theory software package,
which (for better or worse) is protected by a patent.
However it is not hard to obtain access.
- ri: this program computes the individual information for a set
of binding sites.
- makewalker: this program displays sequence walkers
using
ghostscript. I usually use scan and lister (described below) now.
- scan: search a Delila book
with a simple information-theory based weight matrix
to find features.
- multiscan: search a Delila book
several information-theory based weight matricies connected
by variable gaps
to find features.
Examples are
ribosome binding sites
and
sigma70 promoters.
- lister: this program displays sequence walkers
on pages.


Schneider Lab
origin: 1998 February 3
updated: 2020 Dec 30