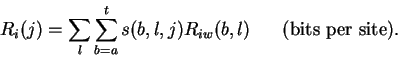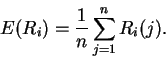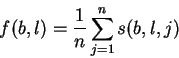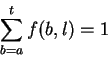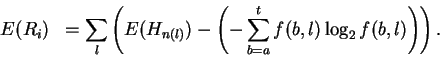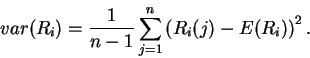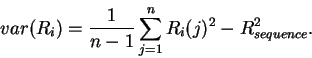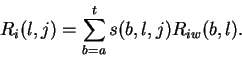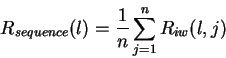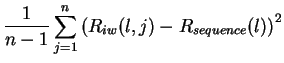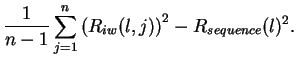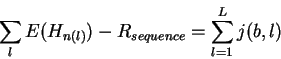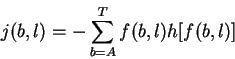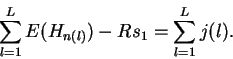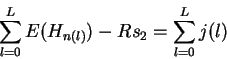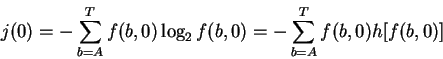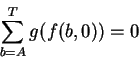Thomas D. Schneider
1
2
with an appendix by John Spouge
3
version = 2.20 of ri.tex 1999 December 23
Journal of Theoretical Biology, 189 (4): 427-441,
1997.
https://alum.mit.edu/www/toms/![]() toms/paper/ri/
toms/paper/ri/
Running title: Information of Individual Sequences
Key words: DNA-protein interactions, information theory, sequence logo, walker
Subject Category: Proteins, Nucleic acids and other biologically important macromolecules: molecular structure.
Related genetic sequences having a common function can be described by Shannon's information measure and depicted graphically by a sequence logo. Though useful for many purposes, sequence logos only show the average sequence conservation, and inferring the conservation for individual sequences is difficult. This limitation is overcome by the individual information (Ri) technique described here. The method begins by generating a weight matrix from the frequencies of each nucleotide or amino acid at each position of the aligned sequences. This matrix is then applied to the sequences themselves to determine the sequence conservation of each individual sequence. The matrix is unique because the average of these assignments is the total sequence conservation, and there is only one way to construct such a matrix. For binding sites on polynucleotides, the weight matrix has a natural cutoff that distinguishes functional sequences from other sequences. Ri values are on an absolute scale measured in bits of information so the conservation of different biological functions can be compared to one another. The matrix can be used to rank-order the sequences, to search for new sequences, to compare sequences to other quantitative data such as binding energy or distance between binding sites, to distinguish mutations from polymorphisms, to design sequences of a given strength, and to detect errors in databases. The Ri method has been used to identify previously undescribed but experimentally verified DNA binding sites. The individual information distribution was determined for E. coli ribosome binding sites, bacterial Fis binding sites, and human donor and acceptor splice junctions, among others. The distributions demonstrate clearly that the consensus sequence is highly unusual, and hence is a poor method to describe naturally occurring binding sites.
Introduction
A flood of sequence data is appearing in the nucleotide sequence databases. To analyze these data, mathematical methods and computer algorithms are needed that are simple, logical and self-consistent. A mathematics that fits these requirements and also connects directly to the physics underlying molecular binding interactions was created by Shannon with the introduction of information theory [Shannon, 1948,Pierce, 1980,Sloane & Wyner, 1993]. Information theory has been successfully used to quantify the sequence conservation in nucleotide and protein sequences [Schneider et al., 1986,Schneider & Stormo, 1989,Eiglmeier et al., 1989,,Penotti, 1991,,,Gutell et al., 1992,Stephens & Schneider, 1992,Papp et al., 1993,,,Pietrokovski, 1996,Blom et al., 1996]. The sequence conservation is given by the average number of bits needed to define a set of aligned sequences. Although this average is useful for understanding the structure of DNA/protein interactions, it does not allow investigation of individual sequences.
This paper describes how the information content of individual sequences can be determined. The method allows direct comparison between the information of particular binding sites to that of other binding sites on the same sequence, to distances between features of the sequence, and to their measured binding energies. It can also be used to search for and to design new binding sites.
Individual information also lends itself to quantitative visualization of complex genetic structures. Previously, only the average picture of a set of binding sites could be depicted graphically by using the sequence logo technique [Schneider & Stephens, 1990]. The individual information method described here is the basis of a new graphic method that shows the information contributed by individual bases in a binding site [Schneider, 1997].
With these tools information theory now provides a common framework for investigating many aspects of genetic sequences.
Theory Individual information of binding sites The information contained in a set of binding sites can be computed by summing the information content across the base positions of the binding sites [Schneider et al., 1986]. But information is an average [Shannon, 1948,Pierce, 1980,Sloane & Wyner, 1993,Schneider, 1995], which suggests that it should be possible to express the average by adding together the information contents of complete individual sequences and then dividing by the number of sequences. This can be done by first creating a weight matrix [Stormo et al., 1982,Schneider et al., 1984,Staden, 1984,Stormo et al., 1986,Stormo, 1990] that assigns an information content to each individual binding site sequence. The matrix is defined so that the average of these values over the entire set of sites is the average information content, as shown below.
The individual information weight matrix is:
In a set of sequences we represent the jth sequence by a matrix s(b,l,j) that contains only 0's and 1's. For example, the sequence 5' CAGGTCTGCA 3'is represented as shown in Fig. 1A. Likewise, an Riw(b,l) matrix for human donor splice junctions is shown in Fig. 1B.
The individual information
of a sequence is the dot product between the sequence
and the weight matrix:
The average information of
the n individual sequences that were used to create the frequency
matrix f(b,l) is the expectation (i.e. mean) of Ri:
Relationship between individual information
and the roots of information theory: surprisal of bases
By expressing formula (6) as a subtraction,
we emphasize that information is a state
function defined as a difference of uncertainties
[Shannon, 1948,Tribus & McIrvine, 1971,Schneider et al., 1986,Penotti, 1990,,Schneider, 1991a,Schneider, 1991b,Schneider, 1994].
The individual information method is consistent with early work
on information theory.
Selecting one symbol from a set of M symbols, requires
no more than ![]() binary decisions [Shannon, 1948].
Rearranging the formula gives:
binary decisions [Shannon, 1948].
Rearranging the formula gives:
The recognition process can be modeled by
the change an individual recognition ``finger'' sees when it
goes from non-specific binding (the before state) to
specific binding (the after state)
[Schneider, 1991a,Schneider, 1994]. In the before
state the average surprisal is 2 bits since there are 4 bases,
while afterwards it will depend on the frequency of the bases
f(b,l) in the binding sites.
The decrease in surprisal is:
Since the individual information is the sum of Riw(b,l) across a binding site, it is the total surprisal decrease from the viewpoint of a particular recognizer binding to a particular sequence. This model allows a recognizer to have different responses to different sequences. Different recognizers have different surprisals for the same sequence because they have different molecular recognition surfaces.
Properties of the individual information distribution The Riw(b,l) matrix can be applied to each sequence that was used to generate the Riw(b,l) itself. A histogram of the number of sites with a given information versus the information displays the Ri distribution (Fig. 2). The expectation of this distribution is by definition Rsequence, the total sequence conservation represented by the area under a sequence logo [Schneider & Stephens, 1990].
According to equation (1), by picking out the most frequent base at each position of the weight matrix, the consensus sequence is assigned the largest Ri value, so the upper bound of the Ri distribution is at the consensus. Likewise, choosing the least frequent base at each position gives the lower bound of the distribution, at the ``anti-consensus''. Since Ri is the sum of a number of small components, its distribution tends to be Gaussian, as dictated by the central limit theorem [Breiman, 1969], assuming that there is only one class of recognizer.
Variance of Ri
Analogous to the mean of the Ri distribution is the spread or
variance of the Ri distribution, given by
Standard error of the mean
By using the Ri distribution,
we can determine the standard deviation of the mean (
Rsequence),
which is known as the standard error of the mean (SEM).
The SEM can be determined directly
from the standard deviation of the Ridistribution (
![]() )
by
)
by
Individual information
variance
at each position in a binding site
Riw(b,l) may also be used to determine the variance at each position
l in the binding site.
First we define the individual information at each position lof each sequence j:
Thermodynamics and individual information
In the case of a molecule binding to a nucleic acid, the
zero coordinate on the Ridistribution can be understood from a thermodynamic viewpoint.
So far, by avoiding the concept of energy when studying pure sequences,
we have avoided making assumptions
about the relationship between information and energy.
That relationship is not a proportionality, it is the inequality
Consider a binding site that has a negative evaluation
by an
Riw(b,l) matrix:
Searches using individual information New sequences can be evaluated and searched for by applying the Riw(b,l) matrix to sequences other than those from which it was derived. Since the numerical value assigned to each position in a sequence by an Riw(b,l) matrix is in bits, the evaluations can be directly compared to the average measures Rsequence and Rfrequency [Schneider et al., 1986].
If a particular base does not appear in the data set used to create
the frequency matrix
f(b,l), then
f(b,l) = 0 and so
![]() at that position
(see equation (1)).
Since there are no known examples of a
functioning site containing the base b at position l,
there is a high degree of surprisal there.
This cannot happen if the matrix is only used to analyze the sequences that
were used to make up the matrix itself
because the infinite positions
are never selected.
(Also, when using the dot product method,
the fact that
at that position
(see equation (1)).
Since there are no known examples of a
functioning site containing the base b at position l,
there is a high degree of surprisal there.
This cannot happen if the matrix is only used to analyze the sequences that
were used to make up the matrix itself
because the infinite positions
are never selected.
(Also, when using the dot product method,
the fact that
![]() ensures that the infinite quantities are suppressed.)
Search programs can handle this situation by replacing
ensures that the infinite quantities are suppressed.)
Search programs can handle this situation by replacing ![]() with
a large negative value.
Alternatively, the search may be relaxed by using a less
severe penalty [Staden, 1984].
The Ri program therefore allows substitution with
1 / (n + t), with
the condition that
with
a large negative value.
Alternatively, the search may be relaxed by using a less
severe penalty [Staden, 1984].
The Ri program therefore allows substitution with
1 / (n + t), with
the condition that
![]() .
For example, using t=1 suggests that the missing base would
be found if just one more binding site sequence were obtained.
However, the ``law of succession of Laplace''
states that
given n trials in which there were k results of one kind,
the best estimate for the probability in another trial
is
(k+1)/(n+2)[Feller, 1968,Papoulis, 1990].
In the present case, we need the probability
of the absence of a particular base when searching for another
binding site, so k = 0 and the best estimate is 1/(n+2).
For this reason we set t=2 for most purposes.
.
For example, using t=1 suggests that the missing base would
be found if just one more binding site sequence were obtained.
However, the ``law of succession of Laplace''
states that
given n trials in which there were k results of one kind,
the best estimate for the probability in another trial
is
(k+1)/(n+2)[Feller, 1968,Papoulis, 1990].
In the present case, we need the probability
of the absence of a particular base when searching for another
binding site, so k = 0 and the best estimate is 1/(n+2).
For this reason we set t=2 for most purposes.
Sampling problems and assumptions It is not possible to determine the information content from a single sequence alone. One reason is that the actual contacts could be anywhere within the sequence, and some positions could be absolutely required (2 bits) while others are completely ignored (0 bits). Without further data, these cannot be distinguished. Another reason is that when frequencies are substituted for probabilities, the information measure becomes biased, and so a small sample correction must be applied [Schneider et al., 1986]. When there is only one sequence the bias is so large that the information content calculated at every position is zero. Yet this paper presents a method for evaluating the sequences of individual binding sites, which may at first appear to be impossible. It is possible because the method is performed in two steps: creating a weight matrix and then evaluating the binding sites with that matrix. There is no contradiction because the individual sites are always evaluated by a model created from a large collection of sequences.
If parts of the sequences are unknown, then the average of the individual information contents generally will not equal the Rsequence as calculated from the frequencies of bases at each position because individual sequences can be strongly affected by missing data. Missing sequences do not affect the overall frequencies much, so Rsequencehardly changes. For this reason calculation of Rsequence should still be done by the original frequencies method [Schneider et al., 1986], and individual information values taken from partial sequence data should be interpreted cautiously.
The individual information method depends on an aligned set of sequences. While multiple alignment is a difficult problem in general, for most binding sites gaps are not required to make good alignments because protein binding sites are generally small objects with little flexibility observed along the sequence. We have recently shown that it is possible to perform rapid gap-free multiple alignment based on information theory [Schneider & Mastronarde, 1996]. A general theory for individual information with gaps is not available, although the uncertainty introduced by gaps has been considered [Schneider et al., 1986] and hidden Markov models may provide the basis for a solution [Krogh et al., 1994].
The model described here assumes that positions along the site are statistically independent from one another. Fortunately, in the cases which have enough sequence samples to be tested, binding sites show almost complete statistical independence. For example, at most 2% of the information in human splice donor sites is in correlations, and none was observed for acceptors [Stephens & Schneider, 1992]. This is also supported by the success of one-layer neural net training (the perceptron) [Stormo et al., 1982,,Brunak et al., 1990a,Brunak et al., 1990b,,Horton & Kanehisa, 1992,Bisant & Maizel, 1995]. Single layer neural networks depend on additivity, and hence their success demonstrates a good degree of positional independence. Furthermore, the closeness of Rsequence and Rfrequencyalso supports independence in a number of cases [Schneider et al., 1986]. In cases that do not show independence, it should be possible to extend the individual information method to account for bases correlated to their neighbors, or even longer relationships [Stephens & Schneider, 1992,Gutell et al., 1992]. However, to do this or to apply it to protein patterns requires many more sequences to avoid the severe effects of small sample size with a large alphabet [Schneider et al., 1986].
Multiple recognizers in a genetic region can affect information theory based models. This problem breaks down into two parts. First, when two or more recognizers have binding sites that are always in the same register with respect to each other, the sequence conservation is higher than expected from the size of the genome and the number of binding sites [Schneider et al., 1986,Herman & Schneider, 1992]. If a thorough information analysis has been done, the situation is easy to detect and in such cases it is unwise to use the individual information matrix because it does not represent a single entity. Second, when nearby sites are not in the same register, the sequence conservation of one site is blurred out in the alignment of the other site. For example, there is no hint of a promoter near the Escherichia coli CRP binding sites [Schneider et al., 1986,Papp et al., 1993].
Results and Discussion Information of individual sequences The first step in individual information analysis of nucleotide binding sites is to gather a number of example sites and to align them using information content as a criterion for good alignment [Schneider et al., 1982,Schneider & Mastronarde, 1996]. After computation of the average information content of the binding sites ( Rsequence) [Schneider et al., 1986] and generation of a sequence logo graphic to inspect the average sequence conservation [Schneider & Stephens, 1990,Schneider, 1996], the aligned sequences are used to generate a model of the binding sites that is called the Riw(b,l) matrix (equation (1)). Because this weight matrix is created from many sequences, it can give statistically significant evaluations of individual sequences, including those used to create the matrix itself. Surprisingly, only one simple criterion is needed to completely determine the weight matrix: it must give individual evaluations to a set of binding sites such that the average of the evaluations is Rsequence.
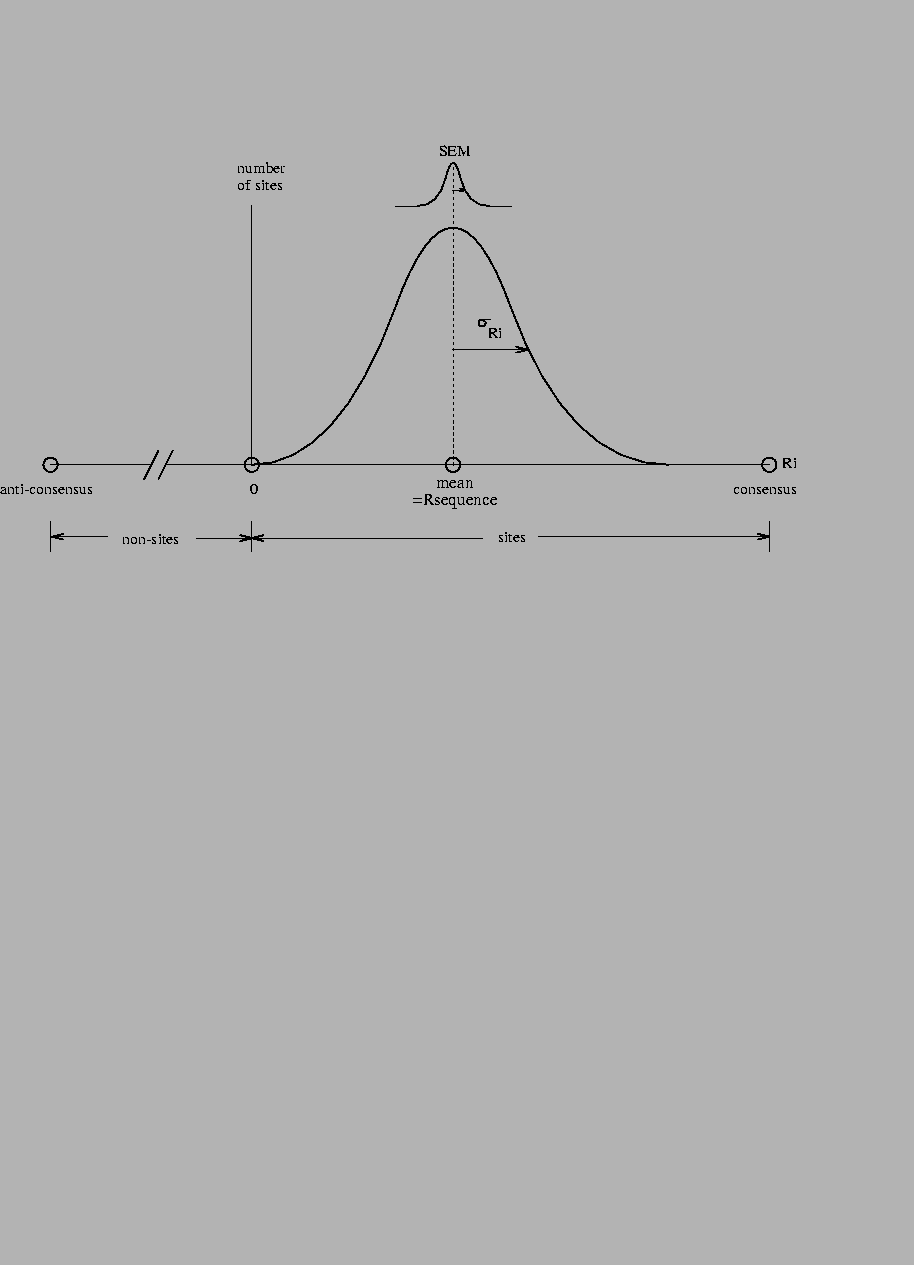
Single binding site conservation distributions The individual conservation distributions for ribosome, donor and acceptor sites are shown in Fig. 3. The majority of the individual information values are above zero (99%, 98%, and 97%, respectively, Fig. 3). This confirms the idea that zero has special significance on the distribution (Fig. 2). A particular sequence might have some parts rated negatively, and other parts rated positively such that the total Ri is zero. These sequences have at best no binding energy according to equation (23), so Ri= 0 classifies sequences into sites and non-sites. Shannon's channel capacity theorem shows that this can be a sharp demarkation [Schneider, 1991a].
Although the distributions are approximately Gaussian, they cannot be exactly Gaussian because the smallest values are truncated at zero. There is also a softer limit at the high end because of the consensus sequence, so the distribution is contained much like a binomial but for practical purposes may be treated as Gaussian.
In rare cases the calculated Ri value is less than zero. This may occur for various reasons. (1) Site sequences may contain sequence or database errors. (2) The Ri is often underestimated when only part of a site's sequence is available. (3) When a limited number of sequences are available to define the distribution, the error for any individual sequence may be appreciable. (4) There may be correlations between parts of the site that are not properly accounted for, although for ribosomes and splice sites these are minimal effects [Stormo et al., 1982,Schneider, 1991a,Stephens & Schneider, 1992]. (5) There may be several kinds of recognizer sites in the data set, an example of which is the new class of splice junctions discussed below.
Identification of distinct classes of sites Hall and Padgett (1994) have observed a new class of splice junctions. Using the acceptor site model developed for Fig. 3, the human acceptor sites in the CMP intron G (GenBank accession M55682, coordinate 396) and P120 intron F (GenBank accession M33132, coordinate 7205) are rated as -3.5 and -3.7 bits respectively. This shows that if the binding sites for several different recognizers have been lumped together, the individual information may be used to help identify the different classes. With enough sequences, multiple classes of sites might be detected by a bimodal or multimodal distribution.
Detecting database errors Like neural networks that have been used to detect errors in a sequence database [Brunak et al., 1990a,Brunak et al., 1990b], negative individual information values have been used to detect errors in data sets for splice junctions, ribosome binding sites and other binding sites. For example, a search of GenBank (72.0 6/15/92) for entries with ``Homo sapiens'' in the source line and ``exon'' in the features gave 4873 entries. The ends of exons were extracted [Schneider et al., 1982] and analyzed with the Riw(b,l) for donor sites from Fig. 1. Of the 6405 exon ends in the 3756 entries that really had exon features, many were not donor sites because many exons end at the poly A site (unfortunately donor and acceptor sites are not explicitly recorded in the database). A large number of exon ends with large negative Ri values were expected (1438 were found), but 842 entries were discovered that had all negative values. An example is the locus HUMEMPB42 (accession M60298) which turned out to be a spliced transcript [Korsgren & Cohen, 1991]. Although portions of the introns are known (figure 2 of that paper) they were not reported to GenBank, only the abutted exons were. (After the error was reported to GenBank, the entry was corrected.)
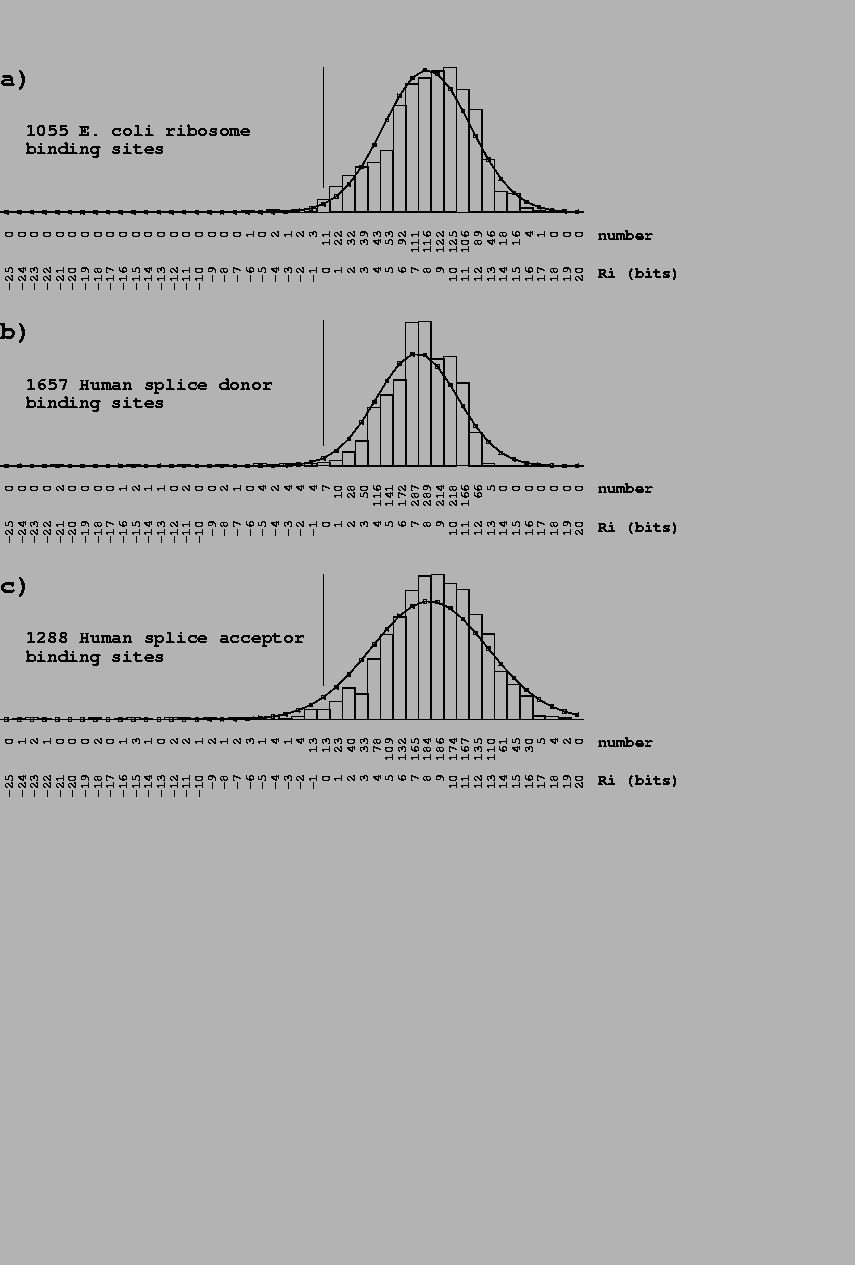
Effect of adding new sites to a binding site model When new sites are added to an individual information model, the evaluation of both the old and new sites changes. Generally this has only a small effect on the old sites once the model has been reasonably well established (Fig. 4). In contrast, new sites almost always increase in value as underrepresented bases become more appropriately represented. On occasion, addition of one site will significantly increase the value of an old site because the new site contains a second example of a base that previously only appeared in the old site.
Correlating binding site conservation with another binding site or a distance The ``exon definition'' model for splicing proposes that the acceptor site is bound first and that the spliceosome then scans downstream across the exon to locate the next donor site [Robberson et al., 1990,Talerico & Berget, 1990,Niwa et al., 1992]. A weak donor might be compensated by a strong acceptor, or the strength of the donor might be related to the distance from the acceptor, so it is important to check whether there are relationships between the donor and acceptor conservation and the exon and intron lengths. Human donor and acceptor splice sites were collected across complete introns and exons, and the individual information of each donor site was plotted against the corresponding acceptor individual information. The Ri site conservations were also plotted against neighboring intron and exon lengths and the total intron-exon interval surrounding each site. No strong correlations were observed (data not shown). A similar lack of correlations between individual splice junctions and each other or with distances between sites across the intron was first noted by F. E. Penotti [Penotti, 1991]. This implies that each human binding site evolves independently to match the spliceosome's molecular surface. Thus Ri can play a role in quantitative analysis of genetic structures.
Correlations of conservation within a single binding site Not only can correlations between whole sites be made, but also correlations between parts of sites can be investigated. A previous analysis of splice junctions suggested that each comes in two parts [Stephens & Schneider, 1992]. To see whether this has an effect on the conservation of these parts, the left half of all donor sites (positions -3 to +1of Fig. 1) was correlated to the right half of the same sites (+2 to +6) giving r = -0.37, and the left half of the acceptor (-25 to -4) was correlated to the right half (-2 to +2) giving r = -0.12. In each case only a weak negative correlation was observed (data not shown), as expected from the requirement for the whole site to have positive Ri.
Strong binding sites are not always natural binding sites Probabilities computed from individual information distributions are curious because sequences with evaluations significantly higher than the mean have low probabilities of being real sites, as can be seen in the distributions (Fig. 3). Strong sites are less likely to appear in the set of natural sites. Evidently the sites evolve to what is required for their function rather than to become the strongest binder. That is, the average of the distribution ( Rsequence) evolves to match the information needed to locate the set of sites in the genome ( Rfrequency) [Schneider et al., 1986,Schneider, 1988,Schneider, 1994].
Consensus sequences are abnormal binding sites Many authors have proposed methods for searching for binding sites in nucleic-acid sequences. The ``consensus sequence'' is widely used by practicing molecular biologists [Day & McMorris, 1992,Prestridge & Stormo, 1993] even though it destroys subtle distinctions in the frequencies of bases in a set of binding sites. This is because choosing the most frequent base at a position is mathematically equivalent to forcing one frequency to 1.0 and all others to 0.0. A glance at some sequence logos [Stephens & Schneider, 1992,Papp et al., 1993] demonstrates that in many binding sites the observed frequencies lie between 0.0 and 1.0 and are not simple fractions such as 1/2 or 1/3.
A consensus sequence is a model of the binding sites. However, to many authors the idea of a consensus sequence has become synonymous with the actual binding sites [Mount et al., 1992,Toledano et al., 1994,Cui et al., 1995]. Thus, for example, it is said that ``the splice site machinery searches a region of the precursor RNA for a consensus 5' splice site'' [Robberson et al., 1990] or ``The splice points are marked by consensus sequences that act as signals for the splicing process'' [Seidel et al., 1992]. The simplest consensus sequence is found by selecting the most frequent base at each position, and therefore by equation (1) gives the largest value obtainable from the Riw matrix. As a result, the consensus sequence lies at the high end of the Ri distribution (Fig. 2). The histograms for ribosomes and splice junctions (Fig. 3) show that most binding sites are not the consensus.
For E. coli ribosomes,
the individual information distribution
over the base range
-21 to +18is characterized reasonably well as
a Gaussian distribution having a
mean and standard deviation of
![]() bits
(Fig. 3).
The consensus
is at 23.98 bits, which is Z = 4.48 standard deviations from the mean,
so the probability of finding such a sequence in wild type E. coli
ribosome binding sites is
bits
(Fig. 3).
The consensus
is at 23.98 bits, which is Z = 4.48 standard deviations from the mean,
so the probability of finding such a sequence in wild type E. coli
ribosome binding sites is
![]() .
No single site (of 1055) was the consensus.
Since there are only about 4300 genes
(GenBank accession U00096), chances are slim that
even one consensus sequence exists in the natural population.
.
No single site (of 1055) was the consensus.
Since there are only about 4300 genes
(GenBank accession U00096), chances are slim that
even one consensus sequence exists in the natural population.
For the compact human
donor sites, over the range -3 to +6,
the mean and standard deviation are
![]() bits with the consensus at 13.13 bits, giving
a Z of 1.6, and a probability of 0.05.
In the set of 1799 sites, only 5 (0.3%) were the consensus.
Even with such a compact binding site, the consensus is not representative
of the whole set.
bits with the consensus at 13.13 bits, giving
a Z of 1.6, and a probability of 0.05.
In the set of 1799 sites, only 5 (0.3%) were the consensus.
Even with such a compact binding site, the consensus is not representative
of the whole set.
Acceptor sites, with the range -25 to +2,
are much more flexible, allowing for a larger consensus.
Their mean and standard deviation are
![]() bits with the consensus at 21.68 bits, giving
a Z of 2.7, and a probability of
bits with the consensus at 21.68 bits, giving
a Z of 2.7, and a probability of
![]() ;
none were in the set of 1744 sites.
;
none were in the set of 1744 sites.
Thus the consensus, rather than being typical, is improbable. If the consensus were the pattern being searched for by a recognizer molecule, as suggested by the statements quoted above, most sites would not be found. One cannot rescue the consensus method by allowing discrete variations such as ``A or G'' [Day & McMorris, 1992] since this still distorts the frequency data. For these reasons, consensus sequences are extremely poor models for binding sites.
Comparison with other quantitative methods
Individual information, although independently derived as described
above, is related to several other methods that use a matrix.
However, important distinctions exist.
Information is the only measure that allows one to
consistently add together ``scores'' from each position in a binding site
[Shannon, 1948],
so other proposed search methods
[Mulligan et al., 1984,Shapiro & Senapathy, 1987,Goodrich et al., 1990,,Bucher, 1990,Quandt et al., 1995]
will give inconsistent results.
The logarithm of probabilities
was proposed as a useful information measure
because it allows addition
of the components, assuming their independence
[Shannon, 1948].
Likewise, various authors have used the natural logarithm of the
base frequencies to create a weight matrix
[Staden, 1984,Berg & von Hippel, 1987,Bucher, 1990,Rice et al., 1992],
but a logarithm alone is not sufficient to identify sites;
some kind of cutoff is required, and usually it is chosen arbitrarily.
For example,
because
Staden's method does not add the factor of 2 bits in equation (1),
all scores are negative with strong ones closest to zero
and so it is not clear where to place a cutoff.
Furthermore, all weights at positions with equiprobable bases would be
assigned ![]() so
the scale shifts depending on the width of the
frequency matrix, and one cannot compare sites
for different recognizers to each other.
Using a consensus to express
a weight matrix evaluation as a percentage of a maximum
[Goodrich et al., 1990,Bucher, 1990,Quandt et al., 1995] also prevents
comparison between recognizers.
Staden's measure also lacks a correction
for small sample size.
Because these sequence evaluation methods lack an absolute scale of measure,
they cannot be used to create a graphic display of binding sites,
such as the walker [Schneider, 1997],
that is consistent for different recognizers.
With natural logarithms the units of the
score are ``nits'',
which have to be divided by
so
the scale shifts depending on the width of the
frequency matrix, and one cannot compare sites
for different recognizers to each other.
Using a consensus to express
a weight matrix evaluation as a percentage of a maximum
[Goodrich et al., 1990,Bucher, 1990,Quandt et al., 1995] also prevents
comparison between recognizers.
Staden's measure also lacks a correction
for small sample size.
Because these sequence evaluation methods lack an absolute scale of measure,
they cannot be used to create a graphic display of binding sites,
such as the walker [Schneider, 1997],
that is consistent for different recognizers.
With natural logarithms the units of the
score are ``nits'',
which have to be divided by
![]() to be directly comparable
to the ``bits'' used in modern
computing and communications systems
[Schneider, 1995].
to be directly comparable
to the ``bits'' used in modern
computing and communications systems
[Schneider, 1995].
The log-odds method, a derivative of the information theory approach [Schneider, 1984,Schneider et al., 1986,Stormo, 1990], does put different kinds of sites on a common scale in bits. However, the average of the log-odds distribution is not the Shannon information content and does not produce a state function [Schneider, 1991b,Schneider, 1994]. It therefore cannot be related to standard definitions of entropy and energy, which are state functions. Further, the log-odds computation of the average can give values larger than 2 bits [Stormo, 1990] even though there are only 4 possible bases. This is because the log-odds method measures the information an observer gains rather than the information gained by the molecular system [Schneider, 1991b]. The states of an external observer are not relevant to molecular interactions, so this computation is not appropriate for the goal of modeling molecular interactions.
The individual information method avoids these various problems by giving an average, consistent with information theory, that allows one to compare different recognizer's sites to each other on an absolute scale given in bits.
The relationship between individual information and
``discrimination energy''
The statistical mechanical approach
to the analysis of binding sequences
assumes that the ratio of the frequencies of bases
is related to the energy by a Boltzmann function
[Berg & von Hippel, 1987,Berg & von Hippel, 1988a,Berg, 1988,Berg & von Hippel, 1988b,,Penotti, 1990,Penotti, 1991].
Strictly following this approach leads to a serious difficulty.
At bacteriophage T7 promoters only half of the 35
bit pattern surrounding the transcriptional start is
required for transcriptional initiation
[Schneider et al., 1986,Schneider, 1988,Schneider & Stormo, 1989].
If the observed patterns actually represent energy
dissipations, then 35 bits worth of energy
is dissipated by the T7 polymerase when it binds.
Yet, experiments show that
the polymerase only requires ![]() bits of sequence pattern
[Schneider & Stormo, 1989].
Since energy must be dissipated to the surroundings
to be useful for molecular binding,
what happened to the ``undissipated'' energy?
How can there be ``discrimination energy'' that is not dissipated
by polymerase binding?
This difficulty can be avoided by referring only to the
information in the sequence patterns:
half of the 35 bit pattern is
used by the polymerase, and the other half is presumably
used by a different recognizer when it binds.
The difficulty with the statistical mechanical approach
stems from
an assumption that energy is equivalent to information.
The Second Law of Thermodynamics
shows that information and energy are related,
but by the inequality in equation (20).
bits of sequence pattern
[Schneider & Stormo, 1989].
Since energy must be dissipated to the surroundings
to be useful for molecular binding,
what happened to the ``undissipated'' energy?
How can there be ``discrimination energy'' that is not dissipated
by polymerase binding?
This difficulty can be avoided by referring only to the
information in the sequence patterns:
half of the 35 bit pattern is
used by the polymerase, and the other half is presumably
used by a different recognizer when it binds.
The difficulty with the statistical mechanical approach
stems from
an assumption that energy is equivalent to information.
The Second Law of Thermodynamics
shows that information and energy are related,
but by the inequality in equation (20).
Associated with the idea of ``discrimination energy'' is a
parameter called ![]() that defines
the relationship between sequence information and
measured binding energies.
that defines
the relationship between sequence information and
measured binding energies.
![]() could be
a function of the position
in the binding site since the information could be closer or further
from its ideal maximum given by
equation
(20).
That is, some binding positions could dissipate more energy than
absolutely necessary
to specify a bit while other positions could dissipate just exactly
the minimum amount.
(An entire binding site
should not be able to
beat the Second Law, but it would be interesting to look
for parts of a binding site that do so by ``coming along for the ride''
as negative weights within functional sites.
Several potential candidates are shown by the upside-down bases of the walker
positioned on the functional site at base 180 in the middle
sequence of
Figure 1 in [Schneider, 1997].
Confirmation would require experimental studies of the binding
energetics of these positions.)
could be
a function of the position
in the binding site since the information could be closer or further
from its ideal maximum given by
equation
(20).
That is, some binding positions could dissipate more energy than
absolutely necessary
to specify a bit while other positions could dissipate just exactly
the minimum amount.
(An entire binding site
should not be able to
beat the Second Law, but it would be interesting to look
for parts of a binding site that do so by ``coming along for the ride''
as negative weights within functional sites.
Several potential candidates are shown by the upside-down bases of the walker
positioned on the functional site at base 180 in the middle
sequence of
Figure 1 in [Schneider, 1997].
Confirmation would require experimental studies of the binding
energetics of these positions.)
The discrimination energy method compares the frequency of a base at
a position in a binding site to the frequency of the consensus base at
the same position [Berg & von Hippel, 1988b,Stormo & Hartzell, 1989].
However, the discrimination energy can easily be calculated from the
Riw(b,l) matrix. Let
![]() be the evaluation of the consensus base at position l,
where ``consensus'' is the most frequent base.
Then, equation (1) gives:
be the evaluation of the consensus base at position l,
where ``consensus'' is the most frequent base.
Then, equation (1) gives:
Individual information compared to training methods With ideal data sets the individual information search method probably cannot give results as good as artificial intelligence methods such as the perceptron [Stormo et al., 1982], neural nets [Nakata et al., 1985,Brunak et al., 1990a,Brunak et al., 1990b,,Horton & Kanehisa, 1992,Bisant & Maizel, 1995], categorical discrimination [Iida, 1987] or hidden Markov models [Krogh et al., 1994] because those methods have the advantage of training on sequences that are not sites.
In practice, however, extensive experimental analysis is needed to avoid contamination of the negative training set by functional sites. In contrast, the information theory method does not require such sites or any cyclic training. As soon as a few experimentally proven sample sites are available, the Riw matrix can be constructed by using an equation. The difficult task of collecting large sets of experimentally demonstrated non-functional sequences is avoided, so there is no concern that one may have contaminated the non-functional examples with real sites [Horton & Kanehisa, 1992]. This lack of training is particularly advantageous for identifying errors for any kind of binding site recorded in a sequence database [Schneider, 1997].
Conclusion The individual information method is simple, but has many useful properties. By this method, individual binding sites can be compared directly to the overall information content, Rsequence, since by definition Rsequence is the average of Ri over the sites. This also allows direct comparison to the predicted average information content given the size of the genome and number of sites ( Rfrequency) [Schneider et al., 1986]. It shows that there is a relationship between the evolution of specific genetic control points and the overall control mechanism in the cell. Individual sequence conservation is measured in standard units, bits, that are easy to manipulate [Schneider, 1995] and allow a wide variety of biological systems to be compared to each other. Because Ri calculations make no assumptions about binding energies, the relationship between energy and information can be investigated experimentally. Applications of the method include the graphical display and engineering of entire genetic control systems [Schneider, 1997] and dissection of binding sites to reveal new kinds of genetic control systems (Hengen et al., in preparation).
Materials and Methods
Programs
Programs of the Delila system
[Schneider et al., 1982,Schneider et al., 1984,Schneider et al., 1986,Schneider & Stephens, 1990]
were used to collect and analyze the sites.
The
Ri program (version 2.37)
generates a
Riw(b,l) matrix and correlates individual sites
with quantitative data.
The
Scan program (version 2.88)
uses the weight matrix to perform searches [Hengen et al., 1997].
It reports the evaluation of each sequence position j in three ways:
as the individual information (Ri(j)),
as the standard deviation from the wild type distribution
mean (
![]() )
and as the one tailed probability (p(j), computed from Z(j)assuming a normal distribution).
The
DNAplot program (version 3.40)
graphs the results in PostScript [Hengen et al., 1997].
Histograms
(Fig. 3)
were generated by the
GenHis program (version 1.73)
written by G. Stormo, and displayed in PostScript by the
GenPic program (version 2.20).
X-Y plots and correlation coefficient computations
(Fig. 4)
were performed by the
Xyplo program (version 8.63).
See http://www-lmmb.ncifcrf.gov/
)
and as the one tailed probability (p(j), computed from Z(j)assuming a normal distribution).
The
DNAplot program (version 3.40)
graphs the results in PostScript [Hengen et al., 1997].
Histograms
(Fig. 3)
were generated by the
GenHis program (version 1.73)
written by G. Stormo, and displayed in PostScript by the
GenPic program (version 2.20).
X-Y plots and correlation coefficient computations
(Fig. 4)
were performed by the
Xyplo program (version 8.63).
See http://www-lmmb.ncifcrf.gov/![]() toms/
for further information about the programs.
toms/
for further information about the programs.
Sequences Ribosome binding sites were from Kenn Rudd's EcoSeq5 database [Rudd & Schneider, 1992]. Human donor and acceptor splice sites were those described in [Stephens & Schneider, 1992]. Fis sites are described in [Hengen et al., 1997].
Acknowledgments I thank Greg Alvord for his help with the statistics, John S. Garavelli for pointing out the mathematics behind the Laplace method, David Lipman for access to GenBank, John Spouge, John S. Garavelli, Kenn Rudd, Paul N. Hengen, Elaine Bucheimer, Denise Rubens, Hugo Martinez, Dong Xu, Stacy Bartram, Kenn Rudd, and especially Pete Rogan for useful conversations and comments on the manuscript, Stacy Bartram for writing DNAscan, and the Frederick Biomedical Supercomputing Center for computer resource support.
John Spouge, National Library of Medicine, Bethesda, MD 20894
First, from equation
(6)
and
E(Ri) = Rsequence,
We reduce the problem further by adding a new position to the range.
Initially the site runs from
![]() and
and
Now we only need to show that at position 0 if
If we define
![]() then showing that g(p) = 0 for all p would finish the proof,
since then
then showing that g(p) = 0 for all p would finish the proof,
since then
![]() except possibly at p=0, but the latter value can be ignored because
of the multiplication by
p = f(b,0) in (34).
except possibly at p=0, but the latter value can be ignored because
of the multiplication by
p = f(b,0) in (34).
We shall insist that h(p) should be continuous,
so the same is true of g(p).
Moreover, equation (34) gives
Because a new base ![]() [Piccirilli et al., 1990]
with frequency zero
(i.e.
[Piccirilli et al., 1990]
with frequency zero
(i.e.
![]() always)
should not affect equation (35),
always)
should not affect equation (35),
![]() .
The frequency vector
.
The frequency vector
Rewriting integer multiplications as repeated additions gives

A) The sequence 5' CAGGTCTGCA 3' represented in matrix format. There is only one ``1'' in each column, marking the base at that position. The remainder of the column is filled with ``0''s. B) The individual weight matrix for human donor splice junctions derived from data given in [Stephens & Schneider, 1992]. The weights of the matrix in B that are selected by the sequence in A are enclosed by boxes. |
The abscissa is the Ri scale in bits, while the ordinate is the number of sites. The Ri distribution is approximately Gaussian. By definition, the mean of the distribution is Rsequence. The standard deviation of the distribution is |
Partially sequenced sites were eliminated from the distributions shown. A. Individual information distribution for 1055 E. coli ribosome binding sites [Rudd & Schneider, 1992]. The mean and standard deviation of the Ri values were fitted by a Gaussian distribution. All sites included the complete range from base -21 to +18of the binding site. A vertical bar marks Ri= 0. B. Sites from a collection of 1799 human donor binding sites [Stephens & Schneider, 1992]. Only 1657 sites that included the complete range from -3 to +6were included. C. Sites from a collection of 1744 human acceptor binding sites [Stephens & Schneider, 1992]. Only 1288 sites that included the complete range from -25 to +2were included. |
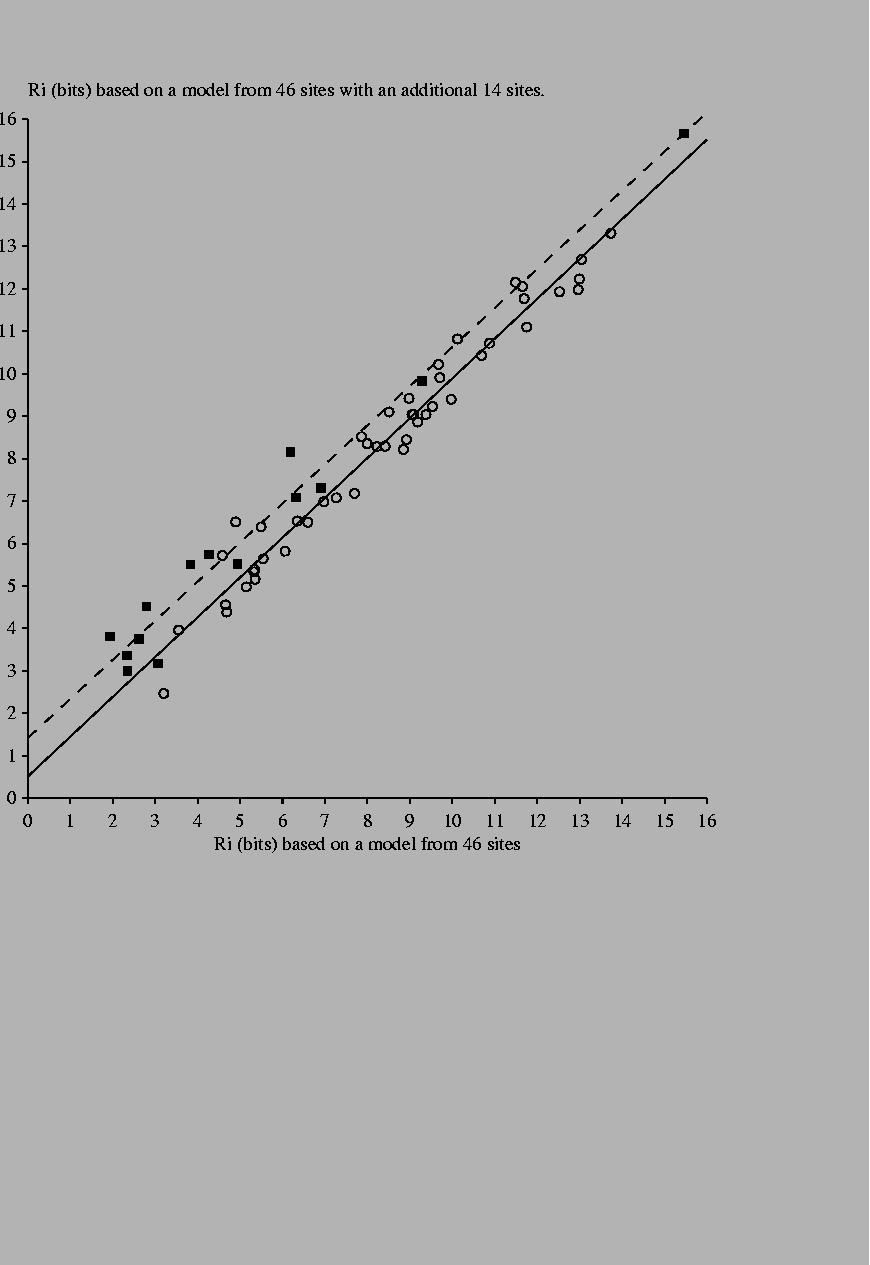
14 new sites [Green et al., 1996,Pan et al., 1996,Falconi et al., 1996] were added to a previous set of 46 Fis binding sites [Hengen et al., 1997]. |