 The Scientific Case Against Intelligent Design
The Scientific Case Against Intelligent Design
The idea that living things are too complex for them to have been developed by evolution is the key concept of the intellectual movement called Intelligent Design. There are two major threads to this movement. Complex Specified Information is purported to measure how unlikely the generation of a biological object is. If the object is highly unlikely compared to a predefined Universal Probability Bound, then Intelligent Design Advocates claim that it could not have evolved. The second thread is that of Irreducible Complexity, in which objects are sought that consist of many interrelated parts. If removal of one of the parts is fatal to the organism (or merely prevents it from reproducing) then Intelligent Design Advocates claim that the system could not have evolved. Both of these concepts are flawed and this is easily and quantitatively demonstrated by a genetic algorithm called Evj that has recently been implemented in Java and can run on any desktop computer.
Evj and its predicessor Ev are computer programs that model the coevolution of DNA binding sites and their recognition protein. Ev was originally written to test the idea that the information in binding sites evolves to match the amount of information required to find the binding sites in the genome. Data from naturally occurring genetic systems shows this correspondence (Schneider1986) and, indeed, this process is well modeled by the Ev program (Schneider.ev2000). You can try this on your own computer using the new Java version of the program, Evj (see the web site schneider.ncifcrf.gov/papers/ev).
The concept of Complex Specified Information was introduced by William A. Dembski in his book No Free Lunch and other places. Basically it says that one can look at a system and compute the amount of information in the system. Dembski has apparently accepted Shannon's information measure for computing Complex Specified Information, as he cited the work on Ev in his book (see Effect of Ties on the Evolution of Information by the Ev program). In addition Dembski introduced the concept of a Universal Probability Bound. This is a probability of the rareness of configuring a physical system. According to No Free Lunch (page 21-22), it is computed from the number of elementary particles in the universe (1080), the maximum rate of physical transitions per second (inverse Planck's constant, 1045 interactions per second), the known age of the universe (13.7 billion years or 4×1017 seconds, Dembski used 1016 seconds), and for good measure an extra factor of a billion for the duration of the universe (109). The Universal Probability Bound suggested by Dembski has a value of 10-150. Dembski argues that a randomly configured system will essentially never have a probability smaller than this Universal Probability Bound:
"Randomly picking 250 proteins and having them all fall among those 500 therefore has probability (500/4,289)250, which has order of magnitude 10-234 and falls considerably below the universal probability bound of 10-150."
-- William Dembski, No Free Lunch: Why Specified Complexity Cannot Be Purchased without Intelligence. Rowman and Littlefield Publishers, Lanham Maryland, 2002. page 293
Dembski applies these two concepts in two steps. First he computes the probability of forming a protein structure (the flagellum in this case) by random assembly from proteins in the cell sequence and finds that it is 10-234. He then notes that this is far less than the Universal Probability Bound. He concludes that the protein could not have formed randomly and therefore it could not have evolved.
There are two fatal flaws to this argument. First, just because something cannot form by a random process does not mean it could not evolve; evolution is not a random process. Dembski is implying that evolution is random but, although mutation is random, selection is not. Secondly, flagella have evolved to self-assemble. Molecular self-assembly was elegantly demonstrated by Bill Wood for the bacteriophage T4 in 1966. It is incorrect to multiply the probabilities of selecting the proteins to form a structure because each protein automatically assembles itself into the structure. The probability of forming a flagellum is close to unity.
Similar inappropriate computations have been made by Lee Spetner (L. M. Spetner, Natural selection: an information-transmission mechanism for evolution, J Theor Biol, 7, 412-429, 1964; Not by Chance: Shattering the Modern Theory of Evolution, 1997, p. 103) and others determine the probability of generating a specific protein sequence. If we chose each of the 20 amino acids at random and assemble them, the probability of generating a protein of 300 amino acids is 20-300 = 10-390.
There is a serious but obvious fatal flaw in this oft-repeated argument. It is that evolution proceeds by small, non-independent incremental steps. Therefore it is inappropriate to compute the probability of protein formation by multiplying the individual probabilities of picking amino acids at random; probabilities can only be multiplied for independent events. While it is true that random joining of amino acids into a polypeptide will not form specific proteins, they can evolve quite easily.
The Evj model demonstrates this in a reasonably easy computation that can be done in less than a day on modern desktop computers. One can set up the program to compute a given amount of information in the binding sites. First, for simplicity, assume that the evolution has proceeded until the information as measured in the sites (Rsequence) has matched that needed to find the sites in the genome (Rfrequency), as is observed in nature in many systems (Schneider1986). This simplifies the problem and it only requires waiting for the evolution to go to completion. That is, we can halt the evolution once all the binding sites are correctly found in the genome and no other positions are found.
So now we can note that
| Rfrequency = log2G/γ, (bits per site) | (1) |
where G is the number of potential binding sites (roughly, the size of the genome) and γ is the number of binding sites. Then the total information in the set of γ binding sites is
|
Rtotal =
γ
×
Rfrequency
= γ × log2G/γ. (bits per genome) |
(2) |
To set up a computer run of the Evj evolution, we need to pick a genome size (G) and the number of organisms in the population. The upper bound of these is determined by the computer memory available. The more organisms we have, the faster they will evolve because lucky discoveries of the next step in evolution are found by individuals.
To maximize Rtotal, should we have more binding sites or less? Having more binding sites would give more total information, but would decrease the information in each binding site because of the inverse relationship between Rfrequency and γ. However, the logarithm function is much 'weaker' than a linear function, so increasing γ increases the total information faster than Rfrequency is decreased. So for a given G we can maximize Rtotal by using as many binding sites as possible. It is wise to avoid overlapping the sites because that might interfere with the evolution.
Given these general conditions, it is easy to set up a simulation.
| population: | 64 creatures |
| genome size, G: | 4096 bases |
| number of sites, γ: | 256 |
| weight width: | 6 bases (standard) |
| site width: | 5 bases |
| mutations per generation: | 1 |
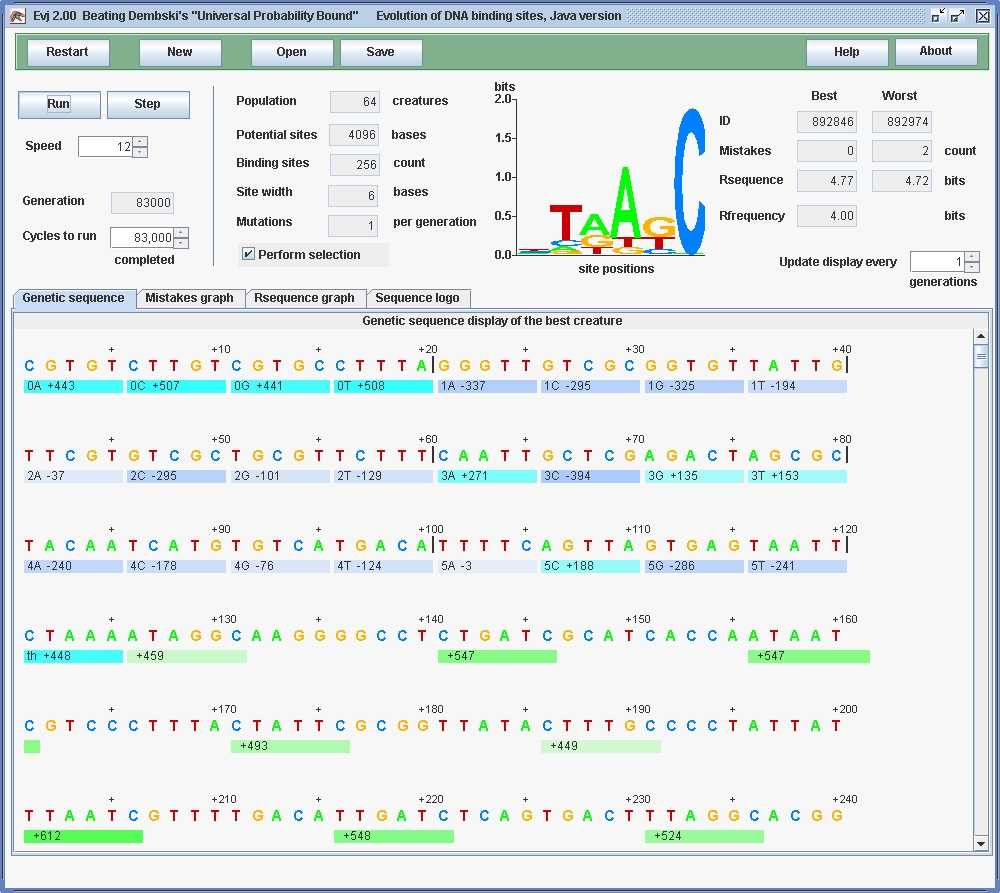
The evolution produced 256 fully functional evolved binding sites; only the first few are shown in the screenshot as green bars under the genomic sequence. (Blue bars mark the gene, further details are given in Experimenting with Evolution: A Guide to Evj.) We know that the sites are all functional because the number of mistakes is zero. The sequence logo, which is the set of colored stacks of letters, shows how the information is distributed across the binding sites by the height of each stack, using a scale of bits (Schneider.Stephens1990). The information is computed using Claude Shannon's information theory (Shannon1948, Pierce1980, Schneider1986). The height of each letter within a stack is proportional to the frequency of that base in the binding sites. You can confirm that the first eight binding sites match the logo quite nicely, but there is variation between the sites.
Although the sites have Rsequence = 4.77 bits, this number fluctuates (which you will see if you run the program) so it is simpler to use Rfrequency to determine what has happened. The program has computed that Rfrequency = log24096/256 = 4.0 bits per site. This is an average, so there is a total of 1024 bits in the 256 sites. The probability of obtaining a 1024 bit pattern from random mutation (without selection of course) is 2-1024 = 5.56×10-309, which beats Dembski's Universal Probability Bound of 10-150 by 158 orders of magnitude! It beats Dembski's protein assembly calculation of 10-234 by 74 orders of magnitude. This was done in less than an hour of evolutionary computation in under 83,000 generations. (A challenge to the reader: keeping the genome size constant, revise the parameters to increase the information 50% more. Prove it by running the model. Answer.)
So what's wrong with Complex Specified Information? Nothing except that we don't need the extra words 'complex' and 'specified', they don't add anything that wasn't in Shannon's original 1948 definition of information, and they only muddy the waters.
What's wrong with Dembski's Universal Probability Bound? Nothing in particular, it is certainly reasonable that one should not expect random events to produce something with a probability less than this number. But it has nothing whatsoever to do with biology because in biology there is evolution by small, sequential and dependent steps. We know this because we find mutations in the laboratory all the time. Some of them cause human diseases (Rogan.Faux.Schneider1998, Khan.Kraemer2002). The evolution of information is amply demonstrated by the Evj program and the reader is encouraged to try it themselves.
Now let us turn to the concept introduced by Michael J. Behe, Irreducible Complexity. This idea was first published in the 1996 book Darwin's Black Box: The Biochemical Challenge to Evolution. The original definition is on page 39 of the book, while a slight revision was recorded in the Kitzmiller v. Dover Area School District court case during Behe's Testimony, day 12:
The underlined words are the change to the text. As far as I can tell, they only make the statement less readable.
Behe's New Definition of Irreducibly Complexity By irreducibly complex, I mean a single system which is necessarily composed of several well-matched, interacting parts that contribute to the basic function, wherein the removal of any one of the parts causes the system to effectively cease functioning.
An irreducibly complex system cannot be produced directly, that is by continuously improving the initial function which continues to work the same mechanisms by slight successive modifications of a pre-cursor system, because any pre-cursor to an irreducibly complex system that is missing a part is, by definition, non-functional.
An irreducibly complex biological system, if there is such a thing, would be a powerful challenge to Darwinian evolution. Since natural selection can only choose systems that are already working, then if a biological system cannot be produced gradually, it would have to arise as an integrated unit in one fell swoop for natural selection to have anything to act on.
Let's apply this to the evolved genomes found by the Evj program.
"a single system": The genetic control system evolved in Evj (and the corresponding ones in nature) comprise a single system.
"which is necessarily composed of several well-matched,": The binding sites match to the recognizer gene as shown by the fact that the organisms make no mistakes.
"interacting parts": The recognizer gene is translated into a weight matrix (representing a binding protein) that binds to the binding sites and therefore interacts with them.
"that contribute to the basic function,": It is critical that this interaction occur for the basic function of the genetic control system.
"wherein the removal of any one of the parts causes the system to effectively cease functioning.": If any binding site is (badly) mutated or if the recognizer gene is modified in a way that it no longer allows recognition of the sites, then the organism makes a lot of mistakes and it is killed in the next selection. One can observe this continuously in the Evj model by watching the 'Worst' mistakes. This number represents organisms that have had mutations that caused them to miss finding one or more binding sites. Sometimes the worst mistakes jumps very high, and this represents failure of the weight matrix. Such organisms die by the next generation.
"An irreducibly complex system cannot be produced directly, that is by continuously improving the initial function which continues to work the same mechanisms by slight successive modifications of a pre-cursor system, because any pre-cursor to an irreducibly complex system that is missing a part is, by definition, non-functional." This is incorrect because the previous generation in the Evj model survives even though the precursor is missing a part. Competition is only with one's peers, not with future generations.
"An irreducibly complex biological system, if there is such a thing, would be a powerful challenge to Darwinian evolution. Since natural selection can only choose systems that are already working, then if a biological system cannot be produced gradually, it would have to arise as an integrated unit in one fell swoop for natural selection to have anything to act on." When the evolution of a system is not understood, Behe is saying that we should give up. If one gives up it merely means that someone else will determine how the system evolved.
These points apply equally well to the results of the Evj program as they do to the observed natural genetic systems that are well modeled by the Evj program, since Rsequence is close to Rfrequency in a number of natural genetic systems.
If I have misapplied the definition, then it is not obvious where. If the definition somehow fails to capture something that would exclude the Evj program (and all known genetic control systems!) that is not obvious either. If there are evolved cases that beat the definition, then we can't just ignore them because they violate the definition. This definition has been around for nearly 10 years, so presumably its author is satisfied with it. Until a revised definition is proposed, we must use it.
So we are forced to conclude that both natural genetic control systems and the Evj model match the definition of Irreducible Complexity at every point. Yet the final Evj genome evolved by mutation, selection and replication in just a few steps, starting from a random sequence. The reader can easily verify this on their own computer using the Java version of the program. This demonstrates that Irreducible Complexity is not a viable concept.
The two fundamental pillars of Intelligent Design, Complex Specified Information / Universal Probability Bound and Irreducible Complexity, were demonstrated to fail given the results of the Evj program.
![]()

Schneider Lab
origin: 2005 Oct 25
updated: version = 1.04 of caseagainstid.html 2006 Jan 18
![]()